





به گزارش سلامت نیوز به نقل از وری ول هلث، سندرم Coffin-Lowry یک اختلال مادرزادی نادر است که با ناتوانی ذهنی، مشکلات قلبی و ویژگیهای مشخص صورت مانند پیشانی برجسته، چشمهای مایل به پایین و بینی کوتاه مشخص میشود.
از آنجایی که سندرم کافین لوری به دلیل جهش کروموزوم X - یکی از دو کروموزوم جنسی ایجاد می شود، علائم بین مردان و زنان متفاوت است. مردان بیشتر تحت تأثیر قرار می گیرند و عموماً علائم بدتری دارند. این اختلال قابل درمان نیست اما قابل مدیریت است. با این حال، مردان مبتلا به سندرم کافین لوری امید به زندگی کوتاه تری دارند.
این مقاله به علائم و علل سندرم Coffin-Lowry میپردازد، از جمله نحوه شناسایی و مدیریت آن و اینکه اگر فرزندتان به این اختلال ژنتیکی نادر مبتلا باشد، چه انتظاری باید داشت.
علائم سندرم کافین لوری چیست؟
علائم سندرم کافین لوری متنوع است، اما با افزایش سن، برجستهتر و مشکلسازتر میشود. این موارد عبارتند از:
- ناتوانی ذهنی: دامنه این علامت از ناتوانی ذهنی خفیف تا عمیق متغیر است، به طوری که برخی از افراد مبتلا به این بیماری هرگز توانایی گفتاری توسعه یافته ندارند.
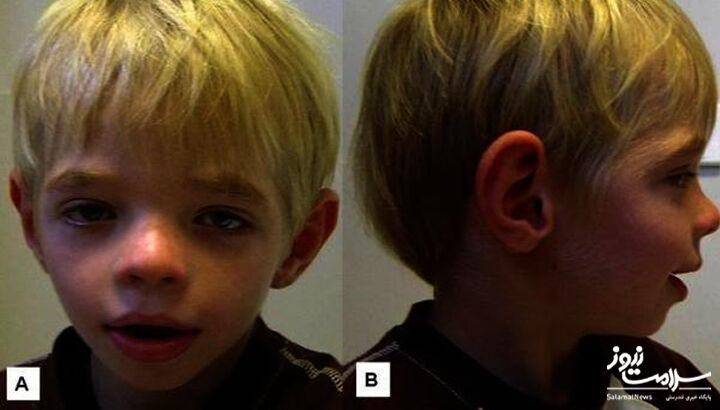
- ویژگی های پهن صورت: به خصوص در مردان برجسته و در اواخر دوران کودکی قابل مشاهده است، افرادی که به سندرم کافین لوری مبتلا هستند، پیشانی برجسته، چشم هایی با فاصله زیاد و کج به سمت پایین، بینی پهن کوتاه و دهانی بازتر با لب های ضخیم تر دارند.
- دستهای بزرگ و نرم: یکی دیگر از ویژگیهای آن، دستهای بزرگتر و نرم با انگشتان کوتاهتر و باریکتر است.
- اپیزودهای افت ناشی از محرک (SIDES): در برخی از افراد در دوران کودکی یا نوجوانی ایجاد می شود، برخی از افراد مبتلا به این وضعیت ممکن است در پاسخ به صداهای بلند یا هیجان فرو بریزند.
- مشکلات قلبی: در طول رشد جنین، کودک ممکن است دچار مشکلات قلبی شود که به اختلال عملکرد دریچه میترال و نارسایی احتقانی قلب کمک می کند.
- مشکلات شنوایی: کم شنوایی حسی عصبی در کودکان مبتلا غیر معمول نیست و شنیدن صدای بلند را سخت می کند و گاهی اوقات منجر به ناشنوایی کامل می شود.
- انحنای ستون فقرات: بسیاری از مبتلایان به سندرم کافین لوری اسکولیوز را تجربه می کنند(انحنای جانبی ستون فقرات) یا کیفوز(گوژپشتی).
- میکروسفالی: یک سر غیرطبیعی کوچک - میکروسفالی- یک علامت مکرر است.
- ناهنجاری های اسکلتی: افراد مبتلا به این سندرم ممکن است دارای مفاصل دوگانه، انگشت شست پا کوتاه شده، استخوان های صورت ضخیم تر، کوتاه شدن استخوان های بلندتر و استخوان سینه نوک تیز یا فرورفته باشند.
- تون عضلانی پایین: شکل گیری ضعیف عضلانی اغلب در افراد مبتلا به سندرم کافین لوری مشاهده می شود.
- اسپاستیسیتی پیشرونده: که به عنوان کشش برخی از گروه های عضلانی تعریف می شود، مشکلی است که می تواند در طول زمان بدتر شود.
- آپنه خواب: یکی از علائم ثبت شده مکرر این وضعیت، آپنه خواب است که در آن هنگام خواب دچار مشکل در تنفس می شوید.
چه چیزی باعث سندرم کافین لوری می شود؟
سندرم کافین لوری یک بیماری مادرزادی است و زمانی رخ می دهد که کودک یک جهش ژنی را در کروموزوم X به نام RPS6KA3 به ارث می برد.
جهش RPS6KA3 باعث کاهش تولید پروتئین عملکردی به نام RSK می شود و درواقع پروتئینی است که نقش مرکزی در رشد و تقسیم سلولی، تخصص سلولی و گردش سلولی دارد. همچنین برای رشد سیستم عصبی مرکزی، به ویژه هیپوکامپ و نئوکورتکس مغز که هر دو در رفتار و شناخت نقش دارند، ضروری است.
بدون RSK کافی، رشد جنین می تواند به خطر بیفتد و منجر به نقص های فیزیکی، فکری و رفتاری شود.
سندرم کافین لوری در یک الگوی غالب مرتبط با X به ارث می رسد، به این معنی که تنها یک ژن جهش یافته برای ایجاد سندرم کافین لوری کافی است. در این الگوی توارث، پدران مبتلا به سندرم Coffin-Lowry نمی توانند این اختلال را به پسران خود منتقل کنند (زیرا پسران یک کروموزوم Y از پدر و یک کروموزوم X از مادر خود دریافت می کنند).
در بین 70 تا 80 درصد موارد، جهش de novo است، به این معنی که از طریق نسلها منتقل نمیشود، اما بدون هیچ دلیل شناختهشدهای خود به خود اتفاق میافتد. 20 تا 30 درصد باقی مانده دارای سابقه خانوادگی سندرم کافین لوری هستند.
آیا سندرم کافین لوری نادر است؟
سندرم کافین لوری یک اختلال ژنتیکی غیرشایع است که از هر 40000 تا 50000 نوزاد یک نفر را مبتلا می کند.
تفاوت بین مردان و زنان
مردان مبتلا به سندرم کافین لوری به مراتب بدتر از زنان دارای ناتوانی ذهنی شدید تا عمیق و تاخیر در رشد هستند. در مقابل، زنان مبتلا به سندرم کافین لوری اغلب عملکرد شناختی طبیعی با ناتوانی ذهنی خفیف، متوسط یا شدید دارند.
دلیل این امر این است که جهش ژن RPS6KA3 در کروموزوم X تا حد زیادی توسط کروموزوم Y فعال می شود و فقط مردان دارای کروموزوم Y (XY) هستند. زنان دارای دو کروموزوم X (XX) هستند.
با این حال، گاهی اوقات زنان دو جهش مختلف را در کروموزوم X به ارث می برند که دیگری می تواند جهش RPS6KA3 را فعال کند. در چنین مواردی، این اختلال می تواند به اندازه مردان شدید باشد.
سندرم کافین لوری چگونه تشخیص داده می شود؟
تشخیص سندرم کافین لوری با ارزیابی ویژگیهای فیزیکی کودک از جمله عملکرد قلب و انحنای ستون فقرات و به کمک تست های تصویربرداری جمجمه و مغز، معمولا با اسکن اشعه ایکس یا تصویربرداری تشدید مغناطیسی (MRI) شروع میشود.
تشخیص را می توان با آزمایش ژنتیک مولکولی تأیید کرد که شامل یک سواب از گونه برای تشخیص وجود و فعالیت RPS6KA3 و RSK2 است.
درصد کمی از کودکان مبتلا به این سندرم هیچ جهش قابل تشخیصی ندارند، اما با این وجود بر اساس علائم بالینی آنها به این اختلال تشخیص داده می شود. وجود یا عدم وجود جهش ژن لزوماً اهداف درمان را تغییر نمی دهد.
آیا سندرم کافین لوری قابل درمان است؟
هیچ درمان یا درمان استانداردی برای سندرم کافین لوری وجود ندارد. درمان بر اساس نوع و شدت علائم کودک است. مراقبت های پزشکی مداوم برای نظارت منظم بر سلامت قلب، شنوایی و سایر عوارض مورد نیاز است.
در صورت نیاز، داروهای ضد صرع ممکن است برای کاهش دوره های افت ناشی از محرک تجویز شود، در حالی که کاشت حلزون ممکن است برای غلبه بر ناشنوایی حسی عصبی توصیه شود.
فیزیوتراپی یا جراحی نیز ممکن است برای درمان انحنای ستون فقرات مورد نیاز باشد زیرا در نهایت میتواند بر تنفس تأثیر بگذارد و مشکلات قلبی را تشدید کند.
امید به زندگی با سندرم کافین لوری
در مردان مبتلا به کافین لوری امید به زندگی کاهش می یابد و تنها تا اواخر دهه 20 زندگی خود زنده می مانند. علت مرگ اغلب مربوط به قلب یا به دلیل عوارض جراحی است. زنان بیشتر زنده میمانند، اما آنها هم نسبت به افراد عادی طول عمر کوتاهتری نیز دارند.
در نهایت، بقا با سندرم کافین لوری بر اساس شدت علائم کودک است، بنابراین هیچ محدودیت مشخصی برای مدت زندگی یا عدم زندگی فرد مبتلا وجود ندارد.
خلاصه
سندرم کافین لوری یک اختلال مادرزادی نادر است که توسط یک ژن جهش یافته در کروموزوم X ایجاد می شود. این وضعیت در مردانی که اغلب ناتوانی ذهنی و جسمی عمیقی دارند، شدیدتر است. سندرم کافین لوری نیز با ویژگی های متمایز صورت، مشکلات قلبی و کاهش شنوایی مشخص می شود.
هیچ درمانی برای سندرم کافین لوری وجود ندارد، اما علائم را می توان به طور موثر مدیریت کرد تا از عوارض جدی مانند نارسایی قلبی جلوگیری کرد. مردان مبتلا به سندرم کافین لوری تا اواخر 20 سالگی زنده می مانند، اگرچه برخی از افراد بسیار طولانی تر زندگی می کنند.








نظر شما